Microfluidics
MICROFLUIDICS. MEGA INSIGHTS.
Get big answers with micro-sized technology
Microfluidics technology empowers you to easily create the approach that’s right for you, so you can find the answers you are looking for and even what you didn’t know was there.
Simplifying your workflow through nanoscale automation maximizes efficiency and provides the flexibility to scale your projects with increased data output. This allows you to adjust your experimental plan to match your needs and your interests with microfluidics-based PCR and NGS library preparation.
Welcome to the power of micro.
NEW FEATURED INSTRUMENT

Biomark X9™ System
for High-Throughput Genomics
Deep Insights With Nanoscale Genomics
The only genomics system for real-time PCR and NGS library preparation to support discovery through screening.


![]()
Miniaturization
Our integrated fluidic circuits (IFCs) have reduced reaction volumes, meaning you use less of your precious samples and reagents.
![]()
Scalability
Scalable sample and target options allow from 144 to 9,216 individual reactions per run without the need to change platforms.
![]()
Flexibility
Precision-manufactured IFCs support different configurations of samples and assays for a broad range of applications and the ability to quickly modify or add to an assay.
![]()
Automation
Automated, streamlined workflows provide the added benefits of time savings, ease of use and standardization.


Download this comprehensive brochure to learn more about our microfluidics instruments, consumables, major application areas and PRO Services.
APPLICATIONS
VIDEOS
Microfluidics Workflow

Learn how enabling a microfluidics-based genomics technology can streamline your lab.
Why Microfluidics?

Learn how your lab can benefit from all the advantages of a genomics-based microfluidics solution.
IFC Loading

See the Nanoflex™ valves at work and take a look at how an IFC automatically distributes and mixes samples and assays in nanoliter volumes, using less sample and less reagent, reducing cost and maximizing output.
HOW IT WORKS
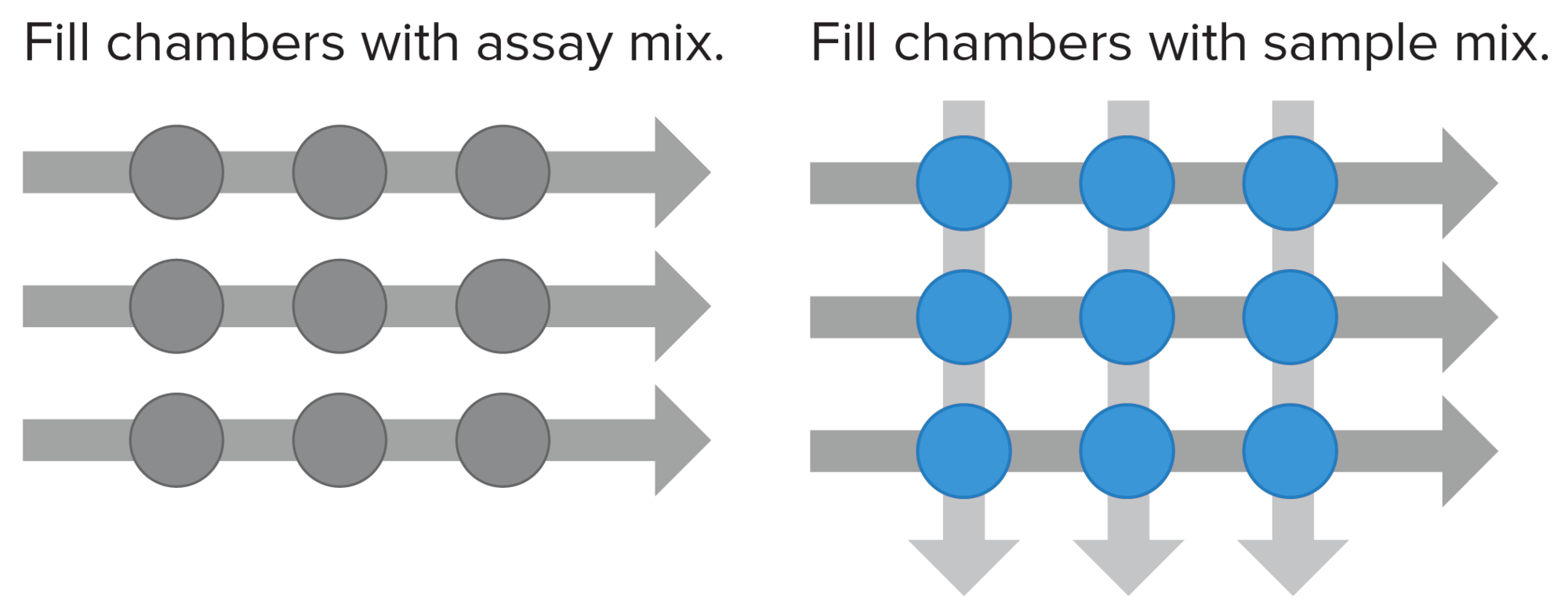
Every Standard BioTools™ IFC contains a network of fluid lines, chambers and Nanoflex valves, rubber membranes that deflect under pressure to pinch off the flow of fluids in a microchannel. The movement, mixing and reactions of samples and reagents are controlled within the chambers of the IFC using microscopic gates, resulting in a closed system that eliminates many individual pipetting steps and has low contamination risk with high reliability. Samples and assays are loaded separately and then combined in a pairwise manner to create unique individual reactions.

WORKFLOW
Design
Easy-to-configure complex panels mean fewer design iterations and more time saved. Our D3™ Assay Design Group can also assist you in creating custom assay panels.
Conserve
About 2 µL of sample can be used for 384 targets, resulting in more than 9,000 reactions and helping you to conserve your precious materials.
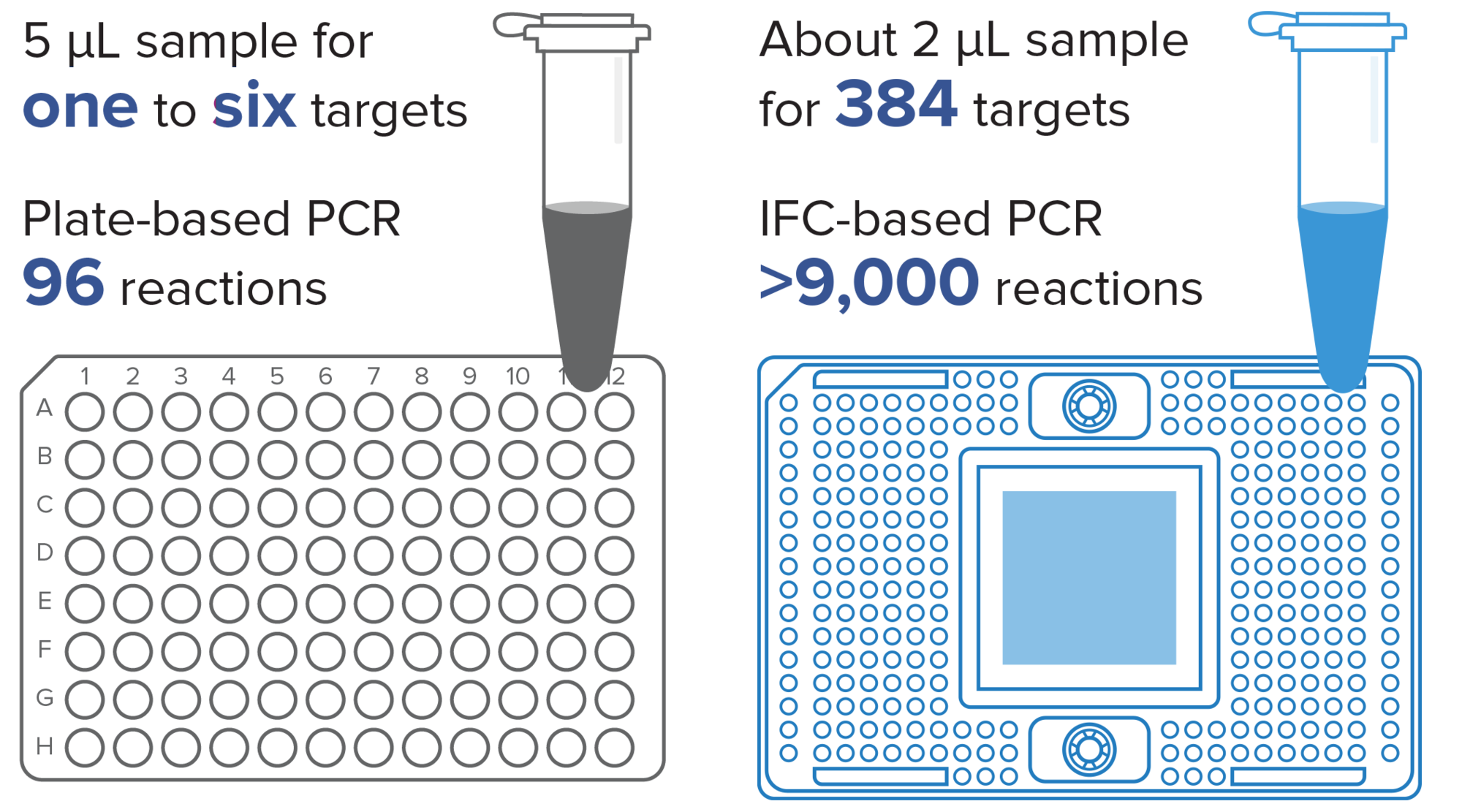
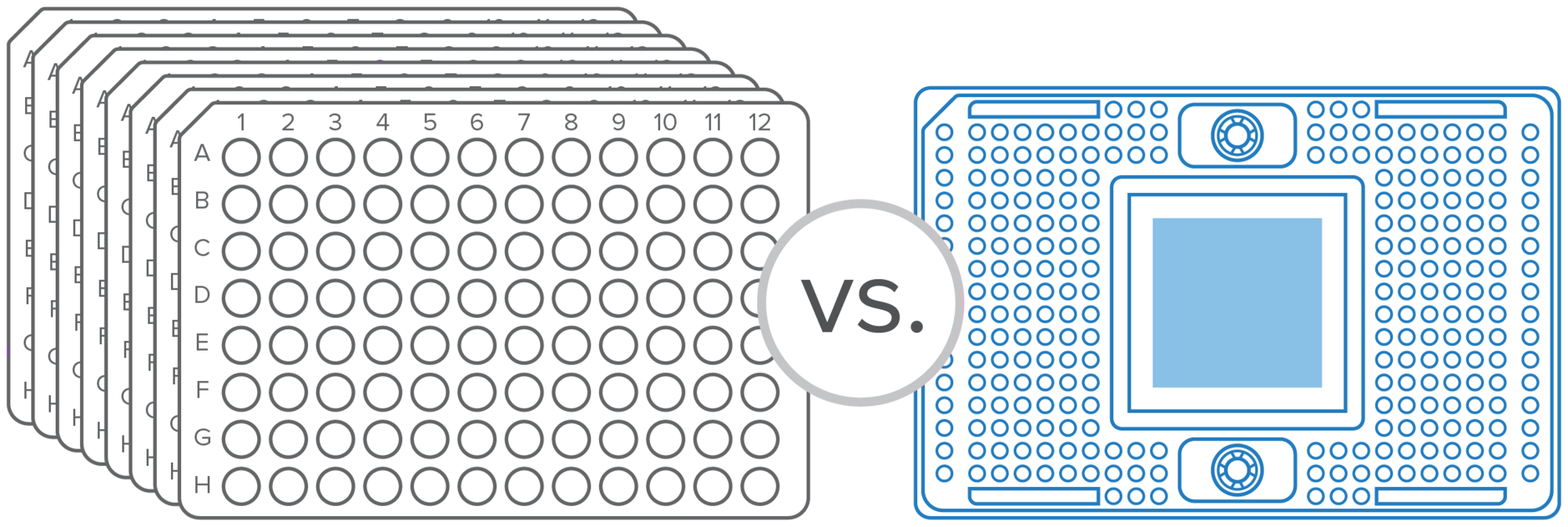
Customize
Thousands of nanoliter-scale inlets help your sample go hundreds of times further than with a 96-well plate. Scale throughput without changing technologies, and add, remove or replace assays on demand.
Analyze
Analyze your results with intuitive software.

WEbinars

What Can Microfluidics Do for You? Nanoscale Automation of Genomics Workflows Now All on One Platform: Biomark X
In this webinar, David King, SVP and Head of Genomics R&D for Standard BioTools, discusses how our newest real-time PCR microfluidics platform simplifies genomic workflows through nanoscale automation to maximize efficiency and cost savings.

Webinar: Genomics Workflows and Applications for the New Standard BioTools Biomark X
In this webinar, Luke Stewart, Director, Field Applications, discusses how liquid-handling robotics can be used in Dynamic Array™ workflows, reviews published real-world application examples and includes Dynamic Array performance data on Biomark X™.

The PCR Advantage: How the Biomark HD System Enhances the TruGraf Gene Expression Test for Kidney Rejection
In this webinar, Juston Weems, PhD, Director of Research and Development for Transplant Genomics, and Standard BioTools will present recently published data on how moving the TruGraf® test to the Biomark HD platform improved specimen collection, assay speed and accuracy, and dramatically improved the sensitivity of the test.
The Standard BioTools Become an Expert: SPARK E-Seminar Series
Register now for this free E-Seminar Series, presented by the Standard BioTools team of scientists. In sessions lasting less than 20 minutes you will learn from our experts and hear how microfluidics technology is being applied around the world to spark breakthroughs in multiple applications.
Events
Join us for one of our upcoming events or watch on demand.
Apr 24 - Apr 25
Location
BSI (British Society of Immunology) Neuro-Immunology Group in Manchester, UK
Apr 25
Location
GW4 Cytomics Symposium in Exeter, UK
Apr 25 - Apr 26
Location
Immuno 2024 in London, UK
May 03
Location
CyTOF Summit | Flow into the Future: Cytometry and Spatial Biology Explored in Edinburgh, Scotland
May 03 - May 07
Location
Immunology 2024 (American Association of Immunologists) in Chicago, IL
May 04 - May 08
Location
CYTO (ISAC International Society for the Advancement of Cytometry) in Edinburgh, Scotland
May 06 - May 09
Location
APHL (Association of Public Health Laboratories) in Milwaukee, WI
May 06 - May 10
Location
WRIB (Workshop on Recent Issues in Bioanalysis) in San Antonio, TX
May 08 - May 10
Location
Precision Med Exhibition and Summit International in Dubai, UAE
May 14 - May 17
Location
EBW Congress (Europe Biobank Week) in Vienna, Austria

Get big answers with micro-sized technology.
View the summaries of the latest microfluidics publications, and learn how customers are making a difference in their labs.
resources
Flyers and brochures
RELATED BLOG POSTS
-
Implementing microfluidics in any lab
No matter what laboratory you work in, using technology that streamlines your processes and saves resources is a crucial part of your success.
Contact us
Interested in Standard BioTools microfluidics products or need a quote? Contact our sales team for product orders or quotes.
Unless explicitly and expressly stated otherwise, all products are provided for Research Use Only, not for use in diagnostic procedures. Find more information here.